
— By Zhenya Senyak
Notes on MPN Mutations, is a description of 15 of the most common driving and passenger MPN mutations currently discovered. These are those genetic changes that create the proteins that play a role in the thrombosis, itching, inflammation and fatigue that characterize the myeloproliferative chaotic blood production system.
The Notes are a guide for MPN patients and caregivers as we stumble into the brave new genomic world that is the cause — and cure – of our disease. The Notes are also a pale tribute to a few of the scientists and technicians whose painstaking work of finding and exploring the MPN mutations is speeding the eradication of this blood cancer.
… classical MPNs and prognosis are determined by a specific genomic landscape, that is, type of MPN driver mutations, association with other mutations, and their order of acquisition. However, factors other than somatic mutations play an important role in disease initiation as well as disease progression such as germ line predisposition, inflammation, and aging. Delineation of these environmental factors will be important to better understand the precise pathenogensis of MPN. (Vainchenker, Kralovics)
Mutation – a force for change
As myeloproliferative neoplasm patients, we’ve come to dread discovering our mutations. We test for signs of mutation with fear and loathing. There’s another side to mutation.
Mutation drives evolution. Evolution proceeds through mutation. Everything changes constantly. And the engine of change (Latin: mutare “to change.”) in biological systems is mutation.
The force that steered us from a future as a puddle of amino acids to a star-trekking, symphony writing organism is mutation.
Some mutations are good for us, help assure our survival. Most mutations, which occur within single cells don’t last very long or are otherwise of little significance. But a few are downright nasty and if they include a survival advantage, they can increase in number and power.
Driver Mutations: A number of lines of evidence support the proposition that mutation in JAK2, CALR or MPL initiate MPN and are sufficient alone to engender a full MPN disease phenotype. (Ciboddo, Mullally)
More than half of patients with PV or ET harbor DNA mutations/variants other than JAK2/CALR/MPL…The presence of some of these mutations adversely affects overall leukemia-free or myelofibrosis-free survival. (Tefferi, et al.)
The Notes can be used to help decipher the nature and impact of those mutations most likely to show up on an MPN patient’s report. (The Myelofibrosis Risk Assessment Tool, a free on-line resource to be released this summer. will incorporate MPN mutational and clinical burdens.) The Grinfeld-Nangalia et al. risk calculator can be found here.
There are many more mutations than those described in the Notes but these 15 lab tested, verified tend to dominate the current landscape.
Mutations naturally arise in the course of cellular reproduction, a complex and decidedly unsexy process of spindles and splitting chromosomes involving transcription to messenger RNA and translation to proteins via ribosomes cruising our cytoplasm before getting folded into a workable, functional proteins. Trillions of cells continually reproducing their yards long alphabet soup of DNA develop inevitable errors – changes — arising from copying, moving, shifting, etc.
Mutations occur within genes, clearly defined relatively short sections of DNA. Sometime they take off expand, usually not. And unless the mutation occurs within the germline – egg and sperm cells –” they are not passed along to our kids. Our genome is made up of two long strips of DNA, one from Mom, one from Dad. If a mutation occurs in both genes on each strip we are homozygous for that mutation.. If it occurs on one gene and not on the other corresponding gene, heterozygous.
A gene is the basic physical and functional unit of heredity. Genes are made up of DNA. Some genes act as instructions to make molecules called proteins. However, many genes do not code for proteins. In humans, genes vary in size from a few hundred DNA bases to more than 2 million bases. The Human Genome Project estimated that humans have between 20,000 and 25,000 genes. ( NIH, US Natl Libr of Medicine.)
Which gets us to allele burden. The percentage of a mutated gene in our genome is the allele burden. So if you’re homozygous for a mutation (mutation on both DNA strands) your maximum allele burden is potentially greater than if your are. heterozygous. (Unmutated genes are called WT or Wild Type.). Zygosity and allele burden have been linked to diagnostic and prognostic conclusions, some debatable.
In the Notes of MPN Mutations each mutation is listed alphabetically to make it easier to locate. We start each description with the location of the mutated gene along its chromosome. The purpose is to help familiarize us with the mutation and illustrate how lengthy a gene may be and how the impact of a particular mutation depends on where along the sprinkling of Letters (bases) the mutation has occurred.
DNA — From here to the Sun
 DNA: the Molecular basis of Mutations…
DNA: the Molecular basis of Mutations…
Mutations are changes in DNA, usually arising during reproduction when copies of DNA have to be translated. DNA is a long string of separate units called nucleotides. “Long” doesn’t actually do DNA justice. Each of us has enough DNA to reach from here to the sun and back more than 300 times.” (Seems unlikely but you can follow the math here if you like.)
In any case, the crowded DNA environment within the cellular nucleus where our mutations arise is staggering. .. 3 billion base pairs of DNA, packaged into 23 chromosomes — times two since most of our 35-50 trillion cells have both Mom and Dad lineages. in each of our nucleated cells!
The composition of DNA: The bases – nucleobases –Adenine (A), Guanine (G) Thymine (T) Cytosine (C) and Uracil (U)) are the nitrogen containing compounds that make up the twisted double helix DNA or RNA.
Technically, genes. a discrete section of DNA, are actually a set of nucelotides in a specific order. Each nucleotide contains a phosphate group, a sugar group and a nitrogen base. The four types of nitrogen bases carried by each nucleotide are adenine (A), thymine (T), guanine (G) and cytosine (C). (G) is always paired with (C) and (A) is paired with (U) or (T). The order of these bases determines DNA’s instructions to produce a string of amino acids that fold up into functional proteins. The nature or function of these protein-coding bases, or codons, can be changed through mutation.
There are several types of mutation. “The 3 MPN-restricted drivers [JAK2, CALR, MPL] lead to a myeloproliferative phenotype. All of the additional mutations in genes involved in epigenetics and splicing modify the differentiation and give a myelodysplastic phenotype even if some of them such as TET2, DNMT3A, and EZH2 clearly play an important role in initiation. Moreover, there are additional mutations involved in splicing that induce myelodysplastic features leading to MF, cytopenia, and eventually progression to leukemia. (Quotation and Illustration: Vinchencker, Kralovics, BLOOD Journal 2017.)
All of the additional mutations in genes involved in epigenetics and splicing modify the differentiation and give a myelodysplastic phenotype even if some of them such as TET2, DNMT3A, and EZH2 clearly play an important role in initiation. Moreover, there are additional mutations involved in splicing that induce myelodysplastic features leading to MF, cytopenia, and eventually progression to leukemia. (Quotation and Illustration: Vinchencker, Kralovics, BLOOD Journal 2017.)
NOTE: Click on illustration to enlarge.
Megakaryocytes (MKs)… beyond platelets –play a central role in MPN pathenogensis
The MPL/JAK2 pathway is activated by the 3 MPN-restricted mutations (JAK2V617F, CALR mutants, and MPL mutants) placing MK hyperplasia and eventually dysplasia as a central determinant in MPN. MKs are mainly involved in platelet production, and also play an important role in the hematopoietic niche by regulating HSCs [hematopoietic stem cells], remodeling the marrow by secretion of TGF-β1 and other cytokines (platelet-derived growth factor [PDGF], vascular endothelial growth factor [VEGF]) which ultimately lead to MF…Furthermore, MKs secrete numerous inflammatory cytokines such as IL1a. The mechanisms of local activation of TGF-b1 are poorly known (Vainchenker. Kralovics)
![]()
The Mutations
![]()
This is an alphabetic listing of 15 of the most prevalent genes based on the Grinfeld, Nangalia study surveying 2005 MPN patients across all MPN phenotypes. Each mutation is ranked, followed by the percentage of patients carrying the mutation. As these figures are derived from a large though limited sample they do not necessarily reflect the actual prevalence of each mutation. Comments on the significance and impact of each mutation are appended to each listing along with a reference to the Source in the Reference Listing at the conclusion of the section.
ASLX1 (Ranking: 3, 21%)
(ASLX Transcriptional regulator)
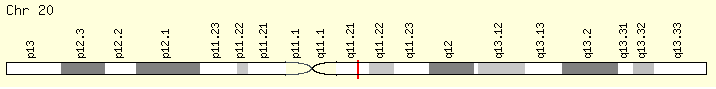
The ASXL1 gene provides instructions for making a protein that is involved in a process known as chromatin remodeling. Chromatin is the complex of DNA and proteins that packages DNA into chromosomes. The ASXL1 protein may have an additional role in gene regulation by signaling to molecules to add a methyl group (a process called methylation) to an area near a gene called the promoter region, which controls gene activity. When a promoter region is methylated, gene activity is repressed.
Another direct means to repress gene activity is to cut the driver mutation out of the cell. This has been done using CRISPR/Cas9 in leukemic cells.
Loss of function mutations in addition-of-sex-combs-like 1 (ASXL1) which encodes a protein that interacts with PRC2 (a complex that governs gene expression through posttranslational modification of histones) are present in approximately 4% of ET, 7 % of PV and 20% of MF patients and area ssoicated with poor prognosis in MF. (Patel)
Among the non phenotypic driver mutations, ASLX1 in particular but also EZH2, IDH1/2 and SRSF2 have a strong impact on prognosis in MF. in which the presence of mutations in any of these genes defines HMR [High Mutational Risk]/ (Ciboddo, Mullally)
The prognostic contribution of mutations other than JAK2, CALR or MPL has been demonstrated in PMF where ASLX1…was identified as unfavorable to survival….Multivariable analysis distinguished CALR(-) ASXL(+) mutational status as the most significant risk factor for survival…for age >65… (Tefferi et al.)
. The ASXL1 mutations are associated with an aggravation in the prognosis, whatever the International Prognostic Scoring System (iPSS) classification. Mutations are loss of function and are associated with a higher frequency of AML transformation. (Vainchenker, Kralovics)
![]()
CALR (Ranking: 2, 32%)
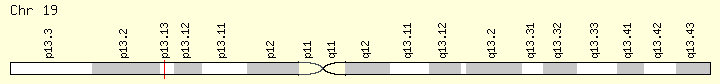
The CALR gene provides instructions for making a multi-functional protein called calreticulin. This protein is found in several parts of the cell, including inside a structure called the endoplasmic reticulum (ER), in the fluid-filled space inside the cell (the cytoplasm), and at the outer surface of the cell. The ER is involved in protein processing and transport, and within this structure, calreticulin plays a role in ensuring the proper folding of newly formed proteins.
The CALR gene provides instructions for making a multi-functional protein called calreticulin. This protein is found in several parts of the cell, including inside a structure called the endoplasmic reticulum (ER), in the fluid-filled space inside the cell (the cytoplasm), and at the outer surface of the cell. The ER is involved in protein processing and transport, and within this structure, calreticulin plays a role in ensuring the proper folding of newly formed proteins. The ER is also a storage location for charged calcium atoms (calcium ions), and calreticulin is involved in maintaining the correct levels of calcium ions in this structure. Through calcium regulation and other mechanisms, calreticulin is thought to play a role in the control of gene activity, cell growth and division (proliferation) and movement (migration), the attachment of cells to one another (adhesion), and regulation of programmed cell death (apoptosis). The function of this protein is important for immune system function and wound healing. https://ghr.nlm.nih.gov/gene/CALR
In conclusion, the 3 MPN oncogenes are true drivers of the disease phenotype with JAK2 exon 12 giving only an erythrocytosis phenotype, JAK2V617F giving rise to ET, PV, and MF, whereas CALR mutant and MPLW515L/K/A are associated with ET and MF, resembling the phenotype observed in patients. The current data sustain the hypothesis that the phenotype of MPNs is mostly related to the types of receptors that are activated. However, in all models, MPL activation plays a central role by its role on HSCs and on megakaryocytes (MKs). (Vainchencker)
The type of MPN phenotypic driver mutation has been shown to have prognostiuc significance in MF. Specifically, a CALR type-1 mutation has been shown to be a good prognostic factor…(.Ciboddo, Mullaly)
Patients with CALR mutations, which co-occurred with LOH at chromosome 19p and with deletion at chromosome 20q, or those with MPL mutations all presented with essential thrombocythemia or myelofibrosis (Grinfeld, Nangalia et al.)
NOTE: Loss of heterozygosity (LOH) is a cross chromosomal event that results in loss of the entire gene and the surrounding chromosomal region. (Wikipedia.)
At the end of 2013, frameshift mutations in the CALR gene were discovered in the majority of JAK22 and MPL2 ET and PMF (50%- 60% ET and 75% PMF).28,29 There are .50 mutations now described, but all are located in exon 9 inducing a 11 (2112) frameshift. At the time of writing, only the mutations leading to this 11 frameshift are known to be pathogenic… . In contrast to wild-type (WT) CALR, the negative charges required for calcium binding are lost at different extents, depending on the mutant. The 2 most frequent mutations correspond to a 52-bp deletion (p.L367fs*46), also called type 1 and a 5-bp insertion (p.K385fs*47), also called type 2. (Vainchencker, Kralovics)
![]()
CBL (Ranking: 10, 6%)
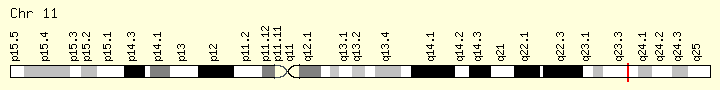
Cbl (named after Casitas B-lineage Lymphoma) is a mammalian gene encoding the protein CBL which is an E3 ubiquitin-protein ligase involved in cell signalling and protein ubiquitination. Mutations to this gene have been implicated in a number of human cancers, particularly acute myeloid leukaemia
Ubiquitination is the process of chemically attaching ubiquitin monomers to a protein, thereby targeting it for degradation. As this is a multi-step process, several different enzymes are involved, the final one being a member of the E3 family of ligases. Cbl functions as an E3 ligase, and therefore is able to catalyse the formation of a covalent bond between ubiquitin and Cbl’s protein substrate – typically a receptor tyrosine kinase.
This gene is a proto-oncogene that encodes a RING finger E3 ubiquitin ligase. The encoded protein is one of the enzymes required for targeting substrates for degradation by the proteasome. This protein mediates the transfer of ubiquitin from ubiquitin conjugating enzymes (E2) to specific substrates. This protein also contains an N-terminal phosphotyrosine binding domain that allows it to interact with numerous tyrosine-phosphorylated substrates and target them for proteasome degradation. As such it functions as a negative regulator of many signal transduction pathways. This gene has been found to be mutated or translocated in many cancers including acute myeloid leukaemia0000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000…. [provided by RefSeq, Jul 2016] https://ghr.nlm.nih.gov/gene/CBL
![]()
DNMT3A (Ranking: 8, 7%)
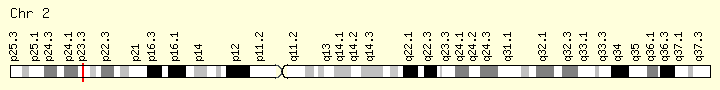
DNA Methyl Transferase 3 Alpha
The DNMT3A gene provides instructions for making an enzyme called DNA methyltransferase 3 alpha. This enzyme is involved in DNA methylation, which is the addition of methyl groups, consisting of one carbon atom and three hydrogen atoms, to DNA molecules. In particular, the enzyme helps add methyl groups to DNA building blocks (nucleotides) called cytosines.
DNA methylation is important in many cellular functions. These include determining whether the instructions in a particular segment of DNA are carried out or suppressed (gene silencing), regulating reactions involving proteins and fats, and controlling the processing of chemicals that relay signals in the nervous system (neurotransmitters). DNA methyltransferase 3 alpha is particularly important for establishing DNA methylation patterns during development before birth. The enzyme also functions in early cells that can give rise to more mature cell types. In early blood cells, called hematopoietic stem cells, the methylation patterns established by DNA methyltransferase 3 alpha promote maturation (differentiation) into different blood cell types.
DNA methylation plays an important role in animal development and gene regulation. In mammals, several genes encoding DNA cytosine methyltransferases have been identified. DNMT1 is constitutively expressed and is required for the maintenance of global methylation after DNA replication
DNA methyltransferases such as DNMT3A attach methyl groups to gene promoter or enhancer sequences suppressing transcripton. DNMT3A mutaitons occur in 15% o f MF patients,7% of PV patients and 3 % of ET patients. (Patel)
Mutations in epigenetic regulators TET2 and DNMT3Aare involved in disease initiation and may precede the acquisition of JAK2V617F. DNMT3A mutations are less frequent than TET2 mutations in MPNs (5%-10%) and in the majority of the cases precede JAK2V617F or MPL mutations. As in other malignant hemopathies, the R882H is the most prevalent DNMT3A mutation. Both TET2 and DNMT3A mutations increase the self-renewal capacities of HSCs in both humans and mice. The precise mechanism is not completely understood, but TET2 and DNMT3A control the expression of genes involved in HSC properties as well as differentiation. It has been recently suggested that TET2-deficient HSCs are more prone to differentiation than normal HSCs, with the derepression of transcription factors such as Klf1.68 TET2 and DNMT3A are the 2 most frequently mutated genes associated with clonal hematopoiesis during aging (Vainchenker. Kralovics)
DNMT3A at 6.8% occurrence is the 8th most common MPN mutation https://ghr.nlm.nih.gov/gene/DNMT3A
![]()
EZH2 (Ranking: 14, 4%)
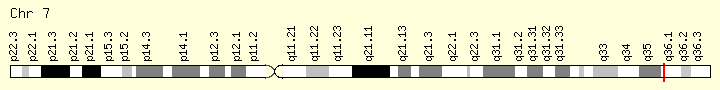
Enhancer of Zeste 2 polycomb repressive complex
The EZH2 gene provides instructions for making a type of enzyme called a histone methyltransferase. Histone methyltransferases modify proteins called histones, which are structural proteins that attach (bind) to DNA and give chromosomes their shape. By adding a molecule called a methyl group to histones (methylation), histone methyltransferases can turn off (suppress) the activity of certain genes, an essential process in normal development. Specifically, the EZH2 enzyme forms part of a protein group called the polycomb repressive complex-2. By turning off particular genes, this complex is involved in the process that determines the type of cell an immature cell will ultimately become (cell fate determination)
Loss of function mutations in EZH2 which enodes the catalytic subunit of PRC2 occur in 13% of MF patients and are associated with loor leukemia free and OS. (Patel).
Loss of EZH2 function promotes MF development and poor prognosis. EZH2 is 1 of the 2 histone methyltransferases of the PRC2 complex… Loss-of-function mutations and cytogenetic lesions in EZH2…have been described in MPNs and across all myeloid malignancies. (Vainchenker, Kralovics)
Among the non phenotypic driver mutations, ASLX1 in particular but also EZH2, IDH1/2 and SRSF2 have a strong impact on prognosis in MF. in which the presence of mutations in any of these genes defines HMR [High Mutational Risk]/ (Ciboddo, Mullally)
![]()
IDH2\DLT3 (Ranking: 11, 5%)

Isocitrate Dehydrogenase 2, Mitochondrial
The IDH2 gene provides instructions for making an enzyme called isocitrate dehydrogenase 2. This enzyme is found in mitochondria, which are the energy-producing centers within cells. Within mitochondria, the enzyme participates in reactions that produce energy for cell activities.
The prognostic contribution of mutations other than JAK2, CALR or MPL has been demonstrated in PMF where …IDH1/2,,was identified as unfavorable to survival. (Tefferi)
Isocitrate dehydrogenase (IDH) gain of function mutations occur in 45 of MF, 2 % of PV and 1% of ET patients IDH1 and IDH2 …are associated with an adverse prognosis in MF…. (Patel).
Among the non phenotypic driver mutations, ASLX1 in particular but also EZH2, IDH1/2 and SRSF2 have a strong impact on prognosis in MF. in which the presence of mutations in any of these genes defines HMR [High Mutational Risk]/ (Ciboddo, Mullally)
In ET [Essential Thrombocythemia] IDH2 and SH2B3 mutations were associated with lower OS, TP53 mutations with lower leukemia free survival and SF3B1 and U2AF1 mutations with higher MF transformation. These findings will need to be validated…(Ciboddo, Mullally)
![]()
JAK2 (Ranking: 1, 66%) https://ghr.nlm.nih.gov/gene/IDH2
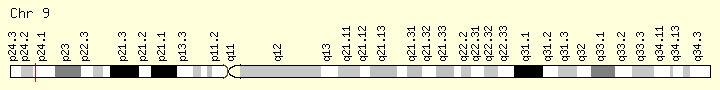
Janus Kinase 2
The JAK2 gene provides instructions for making a protein that promotes the growth and division (proliferation) of cells. This protein is part of a signaling pathway called the JAK/STAT pathway, which transmits chemical signals from outside the cell to the cell’s nucleus. The JAK2 protein is especially important for controlling the production of blood cells from hematopoietic stem cells. These stem cells are located within the bone marrow and have the potential to develop into red blood cells, white blood cells, and platelets https://ghr.nlm.nih.gov/gene/JAK2
In conclusion, the 3 MPN oncogenes are true drivers of the disease phenotype with JAK2 exon 12 giving only an erythrocytosis phenotype, JAK2V617F giving rise to ET, PV, and MF, whereas CALR mutant and MPLW515L/K/A are associated with ET and MF, resembling the phenotype observed in patients. The current data sustain the hypothesis that the phenotype of MPNs is mostly related to the types of receptors that are activated. However, in all models, MPL activation plays a central role by its role on HSCs and on megakaryocytes (MKs). (Vainchenker)’
Patients with JAK2 V617F heterozygosity constituted most of the patients with JAK2-mutated essential thrombocythemia but also some of the patients with polycythemia vera or myelofibrosis; these patients had generally favorable outcomes…The subgroup of patients with JAK2 homozygosity was enriched for patients with NFE2mutations and for patients with polycythemia vera. Myelofibrosis transformations occurred more frequently in this subgroup (Grinfeld, Nangalia, et al.)
IMPACT: Increased RBC, platelet, and granulocyte production. (See: War of the Clones for the “Secrets of JAK2 inhibition therapy persistence.”)
![]()
KMT2A (Ranking: 9, 6%)
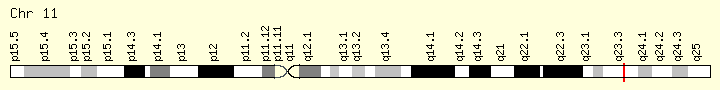
Lysine methyltransferase
This gene encodes a transcriptional coactivator that plays an essential role in regulating gene expression during early development and hematopoiesis. The encoded protein contains multiple conserved functional domains. One of these domains, the SET domain, is responsible for its histone H3 lysine 4 (H3K4) methyltransferase activity which mediates chromatin modifications associated with epigenetic transcriptional activation. This protein is processed by the enzyme Taspase 1 into two fragments, MLL-C and MLL-N. These fragments reassociate and further assemble into different multiprotein complexes that regulate the transcription of specific target genes, including many of the HOX genes. Multiple chromosomal translocations involving this gene are the cause of certain acute lymphoid leukemias and acute myeloid leukemias. Alternate splicing results in multiple transcript variants.[provided by RefSeq, Oct 2010] https://ghr.nlm.nih.gov/gene/KMT2A
![]()
MPL (Ranking: 6, 7%)
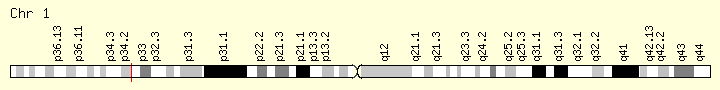
Myeloprofliferative Proto Oncogene Thrombopoietin Receptor
Like essential thrombocythemia, primary myelofibrosis is associated with the MPL gene mutations referred to as W515 mutations. These mutations lead to a constitutively activated thrombopoietin receptor protein, which results in the overproduction of abnormal megakaryocytes. These megakaryocytes stimulate other cells to release collagen, a protein that normally provides structural support for the cells in the bone marrow but causes scar tissue formation in primary myelofibrosis. Because of the fibrosis, the bone marrow cannot produce enough normal blood cells, leading to the signs and symptoms of the condition.
In 1990 an oncogene, v-mpl, was identified from the murine myeloproliferative leukemia virus that was capable of immortalizing bone marrow hematopoietic cells from different lineages. In 1992 the human homologue, named, c-mpl, was cloned. Sequence data revealed that c-mpl encoded a protein that was homologous with members of the hematopoietic receptor superfamily.
Approximately 5% of JAK2 negative cases of ET and 10% of JAK2 negative cases of PMF have activating mutations in the myeloproliferative leukemia virus oncogene (MPL) which encodes the thrombopoietin receptor MPL mutations correlate with lower hemoglobin in ET and ZMF and higher platelet l counts in ET. MPL mutations do not increase risk of thrombosis or fibrotic leukemic transformation, (Patel)
In conclusion, the 3 MPN oncogenes are true drivers of the disease phenotype with JAK2 exon 12 giving only an erythrocytosis phenotype, JAK2V617F giving rise to ET, PV, and MF, whereas CALR mutant and MPLW515L/K/A are associated with ET and MF, resembling the phenotype observed in patients. The current data sustain the hypothesis that the phenotype of MPNs is mostly related to the types of receptors that are activated. However, in all models, MPL activation plays a central role by its role on HSCs and on megakaryocytes (MKs). [Vainchenker.]
Patients with CALR mutations, which co-occurred with LOH at chromosome 19p and with deletion at chromosome 20q, or those with MPL mutations all presented with essential thrombocythemia or myelofibros….Patients with MPL-mutated myelofibrosis had an elevated rate of acute myeloid leukemia transformation (Grinfeld, Nangalia et al.)
![]()
SRSF2 (Ranking: 5, 8%)
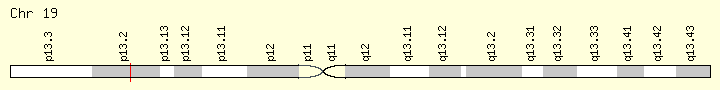
Second Step Splicing Factor 2 ( Peter Pan homolog)
The protein encoded by this gene is a member of the serine/arginine (SR)-rich family of pre-mRNA splicing factors, which constitute part of the spliceosome. Each of these factors contains an RNA recognition motif (RRM) for binding RNA and an RS domain for binding other proteins. The RS domain is rich in serine and arginine residues and facilitates interaction between different SR splicing factors. In addition to being critical for mRNA splicing, the SR proteins have also been shown to be involved in mRNA export from the nucleus and in translation. Two transcript variants encoding the same protein and one non-coding transcript variant have been found for this gene. In addition, a pseudogene of this gene has been found on chromosome 11. [provided by RefSeq, Sep 2010] https://ghr.nlm.nih.gov/gene/SRSF2K]
Loss of function in spliceosome genes that regulate mRNA processing have downstream effects similr to those in loss-of-function muations in cell-cycle regulatory genes and are freuen in MF but are in ET and PV. Mutation in SRSF2 are reported in 17% of MF cases and associated with advanced age, high-risk disese and shorter leukemia free and overall survival. ( Patel)
The prognostic contribution of mutations other than JAK2, CALR or MPL has been demonstrated in PMF where …SRSF2…was identified as unfavorable to survival. (Tefferi)
Among the non phenotypic driver mutations, ASLX1 in particular but also EZH2, IDH1/2 and SRSF2 have a strong impact on prognosis in MF. in which the presence of mutations in any of these genes defines HMR [High Mutational Risk]/ (Ciboddo, Mullally)
SRSF2 mutations are also associated with MPNs having a poor prognosis.93 Mutations in genes encoding spliceosome proteins were detected in different hematopoietic malignancies in 2011, but more particularly in MDS.94 Spliceosome gene mutations in MPNs are essentially restricted to ET and MF. The most frequent mutations include SRSF2, SF3B1, and U2AF1 and are associated with anemia and thrombocytopenia.9
![]()
SF3B1 (Ranking: 7, 7%)
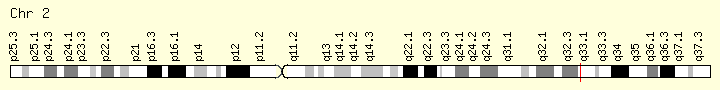
Splicing Factor 3B Subunit 1
Splicing Factor 3B1
The SF3B1 gene provides instructions for making the protein, which is part of a complex called a spliceosome. Spliceosomes help process messenger RNA (mRNA), which is a chemical cousin of DNA that serves as a genetic blueprint for making proteins. The spliceosomes recognize and then remove regions from mRNA molecules that are not used in the blueprint (which are called introns).
Mutations in this gene have been recurrently seen in cases of advanced chronic lymphocytic leukemia,[12] myelodysplastic syndromes[13] and breast cancer.[14] SF3B1 mutations are found in 60%-80% of patients with refractory anemia with ring sideroblasts (RARS; which is a myelodysplastic syndrome) or RARS with thrombocytosis (RARS-T; which is a myelodysplastic syndrome/myeloproliferative neoplasm). There is also an emerging body of evidence to suggest implications of SF3B1 mutations being involved in orbital melanoma. https://ghr.nlm.nih.gov/gene/SF3B1
In ET [Essential Thrombocythemia] IDH2 and SH2B3 mutations were associated with lower OS, TP53 mutations with lower leukemia free survival and SF3B1 and U2AF1 mutations with higher MF transformation. These findings will need to be validated…(Ciboddo, Mullally)
SRSF2 mutations are also associated with MPNs having a poor prognosis.93 Mutations in genes encoding spliceosome proteins were detected in different hematopoietic malignancies in 2011, but more particularly in MDS.94 Spliceosome gene mutations in MPNs are essentially restricted to ET and MF. The most frequent mutations include SRSF2, SF3B1, and U2AF1 and are associated with anemia and thrombocytopenia. (Vainchenker, Kralovics)
![]()
TET 2 (Ranking: 4, 19%)

TET methylcytosine dioxygenase 2
The TET2 gene provides instructions for making a protein whose function is unknown. Based on the function of similar proteins, researchers believe the TET2 protein is involved in regulating the process of transcription, which is the first step in protein production. Although this protein is found throughout the body, it may play a particularly important role in the production of blood cells from hematopoietic stem cells. These stem cells are located within the bone marrow and have the potential to develop into red blood cells, white blood cells, and platelets. The TET2 protein appears to act as a tumor suppressor, which is a protein that prevents cells from growing and dividing in an uncontrolled way.
Although this protein is found throughout the body, it may play a particularly important role in the production of blood cells from hematopoietic stem cells. The TET2 protein appears to act as a tumor suppressor, which is a protein that prevents cells from growing and dividing in an uncontrolled way. https://ghr.nlm.nih.gov/gene/TET2
Mutations in epigenetic regulators TET2 and DNMT3Aare involved in disease initiation and may precede the acquisition of JAK2V617F. DNMT3A mutations are less frequent than TET2 mutations in MPNs (5%-10%) and in the majority of the cases precede JAK2V617F or MPL mutations.As in other malignant hemopathies, the R882H is the most prevalent DNMT3A mutation. Both TET2 and DNMT3A mutations increase the self-renewal capacities of HSCs in both humans and mice. The precise mechanism is not completely understood, but TET2 and DNMT3A control the expression of genes involved in HSC properties as well as differentiation. It has been recently suggested that TET2-deficient HSCs are more prone to differentiation than normal HSCs, with the derepression of transcription factors such as Klf1.68 TET2 and DNMT3A are the 2 most frequently mutated genes associated with clonal hematopoiesis during aging (Vainchenker. Kralovics)
![]()
TP53 Genomic Sub=group #1 (Ranking:19, 2%)
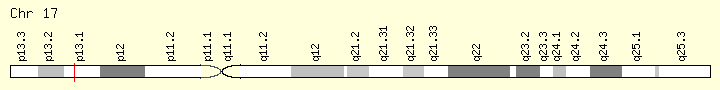
Tumor Protein P53
The TP53 gene provides instructions for making a protein called tumor protein p53 (or p53). This protein acts as a tumor suppressor, which means that it regulates cell division by keeping cells from growing and dividing (proliferating) too fast or in an uncontrolled way.
The p53 protein is located in the nucleus of cells throughout the body, where it attaches (binds) directly to DNA. When the DNA in a cell becomes damaged by agents such as toxic chemicals, radiation, or ultraviolet (UV) rays from sunlight, this protein plays a critical role in determining whether the DNA will be repaired or the damaged cell will self-destruct (undergo apoptosis). If the DNA can be repaired, p53 activates other genes to fix the damage. If the DNA cannot be repaired, this protein prevents the cell from dividing and signals it to undergo apoptosis. By stopping cells with mutated or damaged DNA from dividing, p53 helps prevent the development of tumors.
Because p53 is essential for regulating cell division and preventing tumor formation, it has been nicknamed the “guardian of the genome.”
In ET [Essential Thrombocythemia] IDH2 and SH2B3 mutations were associated with lower OS, TP53 mutations with lower leukemia free survival and SF3B1 and U2AF1 mutations with higher MF transformation. These findings will need to be validated…(Ciboddo, Mullally)
TP53 mutations, often co-occurring with aberrations at chromosome 17p, and deletions at chromosome 5q identified the first subgroup. TP53 mutations often occur later in disease but dominate the genomic and clinical features of these patients regardless of the initial driver of the myeloproliferative neoplasm. As in patients with other blood cancers with TP53 mutations,these patients have a dismal prognosis with a high risk of transformation to acute myeloid leukemia …. Of the eight subgroups of myeloproliferative neoplasms identified, the subgroup with TP53 mutations was genomically unstable and had poor outcomes; this same subgroup, with similar clinical implications, has been identified in acute myeloid leukemia and other hematologic cancers (Grinfeld, Nangalia et al.)
Various mutations and cytogenetic lesions targeting the TP53 tumorsuppressor function are found in around 40% to 50% of secondary leukemia of MPNs. This usually includes missense mutations or deletions in the TP53 gene or amplification of chromosome 1q targeting MDM4, a TP53 transcriptional inhibitor.98 Heterozygous TP53- mutated clones can be found early in MPN, but evolution to leukemia is associated with its clonal dominance and transition from heterozygosityto homozygosity forthe mutation. (Vainchenker, Kralovics)
Cytogenetic Location: 17p13.1, which is the short (p) arm of chromosome 17 at position 13.1. Molecular Location: base pairs 7,668,402 to 7,687,550 on chromosome 17 (Homo sapiens Annotation Release 109, GRCh38.p12) (NCBI)
U2AF1 (Ranking: 13, 4%)
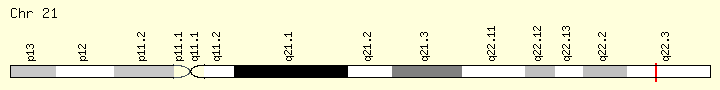
Small Nuclear RNA Auxiliary Factor
This gene belongs to the splicing factor SR family of genes. U2 auxiliary factor, comprising a large and a small subunit, is a non-snRNP protein required for the binding of U2 snRNP to the pre-mRNA branch site. This gene encodes the small subunit which plays a critical role in both constitutive and enhancer-dependent RNA splicing by directly mediating interactions between the large subunit and proteins bound to the enhancers. Alternatively spliced transcript variants encoding different isoforms have been identified. [provided by RefSeq, Jul 2008]
Loss of function in spliceosome genes that regulate mRNA processing hve downstream effects similr to those in loss-of-function muations in cell-cycle regulatory genes and are freuen in MF but are in ET and PV. Mutations in U2AF1 are reported in 15% of PMF cases and associated with anemia and thrombocytopenia. ( Patel)
In ET [Essential Thrombocythemia] IDH2 and SH2B3 mutations were associated with lower OS, TP53 mutations with lower leukemia free survival and SF3B1 and U2AF1 mutations with higher MF transformation. These findings will need to be validated…(Ciboddo, Mullally)
USAF1, with 3.9 % occurrence is the 13th most common MPN mutation.
![]()
The Practical Implications of Genomics
It is hard to predict the ultimate therapeutic power available in fusing genetic engineering, gene editing, and immunotherapy. It is possible now to correct some blood cancers and edit out point mutations. But whether or not – or when – we develop the reliable capability to create biochemical entities and delivery systems capable of seeking out and destroying MPN mutant clones, the primary value of genomic testing right now is in prognostic.
The Power of Prognosis
Because the current standard of medical practice is to assess and treat the MPN phenotype, the result of genomic instability, and not the cause — mutation and chromosomal anomalies — our ability to predict the nature and course of an MPN is limited.
That approach leaves us with stark, almost binary, choices. We can treat or simply observe chronic MPNs, low risk ET and PV, or aggressively treat higher risk MPNs. The issue is specially critical in Intermediate risk assessments, . For higher risk patients we still only have meds that temporarily improve symptoms by inhibiting the JAK2 mutation. It is a potentially lethal option, masking the expansion of disease and the likely piling on of newly arising collaborating mutations in the march toward AML.
The practical implications of genomic testing, the value of identifying the number, nature, and timing of mutations and chromosomal changes is the gaining of new clarity in understanding both the current disease status and its likely progression.
Instead of treating a generic phenotype like ET,PV, and MF we gain the option to target the mutations themselves and the known outcome of their unrestricted expansion. The old MPN categories are useful but crude. MF needs a questionable Pre-MF category to account for certain clinical developments and its progress toward acute myeloid leukemia cannot be predicted with certainty. PV can often not be distinguished from ET without red cell mass testing and sometime not even then. Intermediate MF risk levels are subject to debate. But an ASLX1 mutation, a TET2 mutation can be identified and timed and an existing personal mutational landscape fully mapped to determine the prognosis and drive the medical intervention choices.
When the ultimate choice is stem cell transplant, swapping in a new, healthy hematopoietic system, the odds soar in our favor when we exercise that option earlier, rather than later. No one wants to start the SCT procedure prematurely or when it might not be necessary. But it is that prognostic uncertaintly that has resulted in the deaths of so many MPN patients. An uncertainty that can be reduced through genomic testing.
Grinberg, Nantalia et al finding “The inclusion of mutations and chromosomal changes beyond JAK2, CALR, and MPL improved the predictive power of prognostic models.: have “implemented a free, user-friendly online calculator of individualized patient outcomes..” The calculator can be used to explore data from the “Classification…” cohort or, more personal input. You can access it here: https://cancer.sanger.ac.uk/mpn-multistage
. (We had no luck since we got “Disconnected from Server” each time we tried to calculate risk but there are tabs on that the Calculator worth visitng.) The GENOMIC tab lets the viewer page down through major mutations and karyotype anomalies and breaks down, graphically the percentage of ET, PV or MF patients carrying that mutation. Try it and maybe demonstrate it for your hematologist.
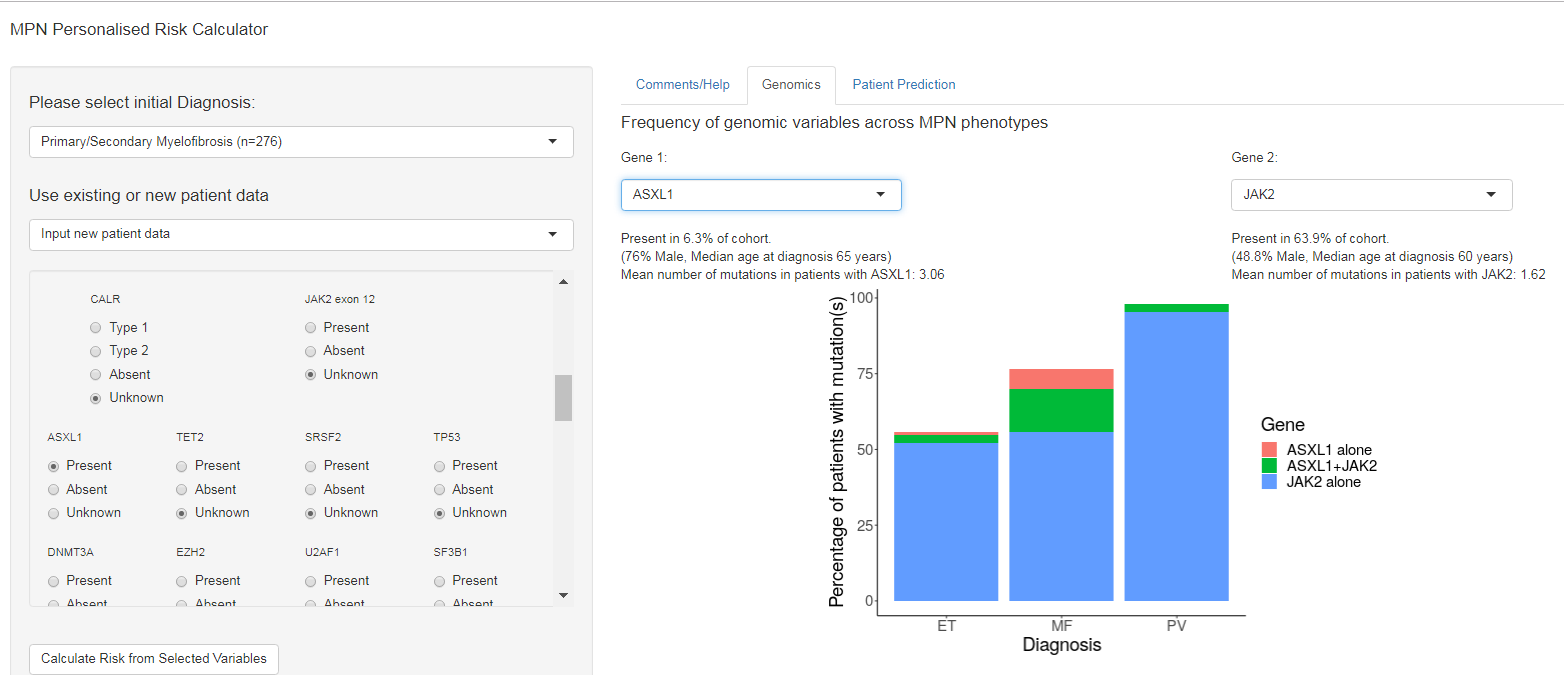
![]()
Legal Risks
Despite DNA testing upending the practice of medicine, a current MPNforum patient survey revealed the great majority of MPN physicians (>80 %) do not routinely order genomic tests for their patients. That may no longer be prudent.
According to Science (10 May 2019, “Genomics breeds new legal questions“) “Failing to offer such tests carries legal risks of its own.” In Pennsylvania, a judge ruled a physician who failed to order genetic testing after his patient’s EKG indicated an underlying genetic disease should have alerted the family of the teenage son whose life might have been saved had they known of his father’s genetic anomaly.
The legal issues extend to cases where knowledge of the impact of known mutations expands. What is the physician’s responsibility to contact a patient with reinterpretation of a lab’s results? What’s the actual risk> “(Gary) Marchant a legal law professor…and his colleague Rachel Lindor …an emergency medicine physician at the Mayo CLinic in Phoenix, unearthed 202 genomic malpractice cases…About 60% of genomics cases resulted in a pyout to the plaintiff, they found, compared with at most 22% of those in other medical malpractice areas.”
![]()
References
Here are sources cited in the Notes available to consult on-line for more detailed information. Each of these publications has its own rich bibliography.
Ciboddo, Mullaly, ASH education book, 2018… Jak2 (and other genes) be nimble with MPN diagnosis….pp 110-115
Grinfeld, Nangala et al., NEJM, Ovtober, 2018,.. Classification and Personalized Prognosis in Myeloproliferative Neoplasms.
Patel, A, et al. Clinical Cancer Research, 2016, New strategies in myeloproliferative neoplasms, the evolving genetic and therapeutic landscape.
Schuschlik, F, Kralovics, R. Expert Rev Hematol 2017, Mutations in myeloproliferative neoplasms – their significance and clinical use
Tefferi, A et al., Leukemia,2014…CALR and ASXL1 mutations-based molecular prognositication in primary myelofibrosis…
Tefferi, A et al., Blood advances, Nov, 2016, Targeted deep sequencing in polycythemia vera and essential thrombocythyemia.
Vainchenker, Kralovics, Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms., 2017, BLOOD, Ash Review Series.
Valletta, S et al., Oncotarget, 2015…ASLX1 mutation correction by CRISPR/Cas9 restures gene function…
and: the NIH National Library of Medicine, Genetics Home Reference
© 2019, MPNforum. All rights reserved under Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License